The dilemma about computationally inferred function annotation
The
Gene Ontology initative is the standard for protein function annotation. For 2011 alone, Google Scholar finds almost 10,000 scientific articles with the keyword "Gene Ontology".
The trouble is that we know little about the quality of these annotations, especially the >98% inferred computationally. The community perceives them as unreliable--at best suited for relatively coarse exploratory analyses, such as term
enrichment analyses (and even those are
not without risks).
At the same time, virtually everything we know about the function of genes in non-model organisms is based on computational function inference.
Our approach: verify old, computationally-inferred annotations using new experimentally established annotations
Nives Škunca, first author of our study, came up with the fundamental idea: to use experimentally-backed annotations, considered the gold standard, to verify computational ("electronic") annotations. And to avoid circularity, we made sure to only use experimental annotations added to the GO database (
UniProt-GOA, to be precise)
after the computational annotations under evaluation.
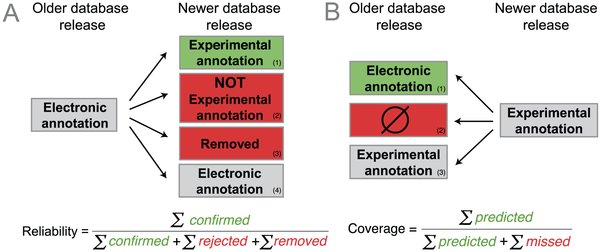
Based on this idea, we defined the average
reliability of a GO term as the proportion of electronic terms in an older database release confirmed by new experimental annotations in a subsequent release (see figure below). Hence, every time a new experimental annotation confirms an electronic prediction, the reliability of the corresponding term increases. Conversely, every time a new experimental annotation contradicts an electronic annotation or, more subtly, every time an electronic annotation is subsequently removed from the database, the reliability of that term decreases. Our reliability measures attempts to capture the
machine learning notion of precision.
To capture the
machine learning notion of recall, we defined the
coverage measure, the fraction of new experimental annotations computationally predicted (see figure above). For instance, a high coverage means that most new experimental annotation has been previously predicted as electronic annotation.
Reliability measure: not as straightforward as it might seem
At first sight, these definitions might seem quite mundane. But let's have a closer look at the reliability measure, which proved to be much more tricky--even contentious, see next section--to devise than we had anticipated.
The open world assumption makes it difficult to falsify predictions. Consider a computational prediction assigning function x to a certain gene. If an experiment later demonstrates that this gene has function y, this does not imply that the original prediction was wrong. What we need to falsify the prediction is an experiment that demonstrates that gene does not have function x.
Such "negative results" can be captured in GO annotations
using the NOT qualifier. But a
search on EBI QuickGO reveals that <1% of current experimental annotations are negative annotations. In part, this state of affairs is a consequence of the general bias against negative results in the literature. Also, it is harder to make definitive statements about absence of function than about presence of function, as absence must be ascertained under all relevant conditions.
Reliability measure: penalising removed annotations
If you recall our definitions above, we include in the reliability measure a penalty for electronic annotations that are subsequently removed from the database (an annotation present in release n has disappeared in release n+1).
However, electronic annotations can disappear for reasons other than being wrong. As Emily Dimmer and colleagues from UniProt-GOA pointed out to us, removals can reflect tightening standards (e.g. by setting more conservative inference thresholds), responses to changes in the
GO structure, or temporary omissions due to technical problems (e.g. integration failure from external resources).
Nevertheless, we reasoned that from the standpoint of a user, removed annotations do not inspire confidence and whatever the reason may be, removed annotations can hardly be considered "reliable".
This discussion also highlights the importance of finding an appropriate name. Because a removal does not necessarily implies an error, calling our measure "correctness" or "accuracy" would have been too strong. Conversely, calling our measure "stability" would also not have been appropriate, as it goes beyond mere stability: electronic annotations that are left unchanged do not increase the reliability ratio of a term; only experimental confirmation does.
What we found
One main finding of our study is that electronic annotations have significantly improved in recent years. A way of seeing this is to look at the following interactive motion plot (click on image to load the flash app):
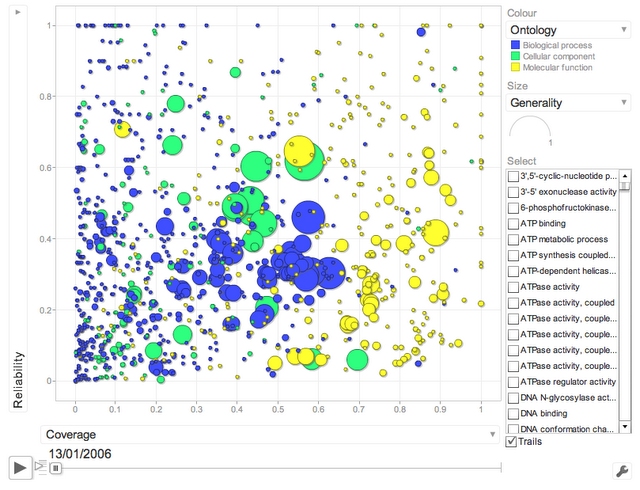
(click on image to load the flash applet)
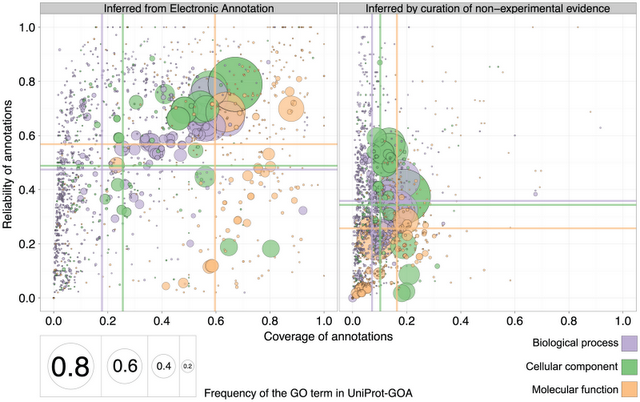
Better yet, we also observed that the reliability of electronic annotations is even higher than that of annotations inferred by curators (i.e. when they use evidence other than experiments from primary literature):
Looking forward, we view this work as an essential step toward our long-term aim of improving computational function inference. Indeed, one thing that seems to often hold in computational biology is that there is no point coming up with a faster or more clever algorithm as long as one has not identified a dependable objective function (or assessment strategy), such as the quality measures introduced here. As late management-guru Peter Drucker said, "
there is nothing so useless as doing efficiently that which should not be done at all".
Reference:
Skunca Nives, Altenhoff Adrian, & Dessimoz Christophe (2012). Quality of computationally inferred gene ontology annotations. PLoS computational biology, 8 (5) PMID: 22693439


{kind=link}
Recent Comments